Here’s a clear, engaging, and medically accurate English educational article on Prader–Willi Syndrome (PWS), written in the same structured style as your previous requests — suitable for teaching, publication, or public health education.
Prader–Willi Syndrome: A Genetic Disorder of Growth, Appetite, and Hormones
Prader–Willi syndrome (PWS) is a rare genetic disorder affecting multiple body systems. It is characterized by hypotonia (low muscle tone) in infancy, followed by hyperphagia (excessive appetite) leading to obesity, along with developmental delay, short stature, and hormonal deficiencies.
PWS results from the loss of function of paternally expressed genes on chromosome 15q11–q13, and is one of the most common causes of genetic obesity.
1. Historical Background and Eponym
The syndrome was first described in 1956 by Swiss physicians Andrea Prader, Heinrich Willi, and Alexis Labhart, who reported a cluster of features:
short stature, hypotonia, hypogonadism, obesity, and cognitive impairment.
It was later named Prader–Willi syndrome in honor of its primary discoverers, with modern genetics identifying its chromosomal origin.
2. Genetic and Pathophysiological Basis
Cause:
PWS results from loss of paternally expressed genes in the 15q11–q13 region of chromosome 15.
Mechanisms (three main genetic causes):
Paternal deletion (≈ 70% of cases): deletion in the paternal 15q11–q13 region
Maternal uniparental disomy (UPD) (≈ 25%): both copies of chromosome 15 come from the mother
Imprinting defects or translocations (≈ 1–5%): errors in gene expression control
Key concept:
This region contains imprinted genes—active only when inherited from the father. Loss of these paternal genes disrupts hypothalamic regulation, leading to hormone imbalance, appetite dysregulation, and growth abnormalities.
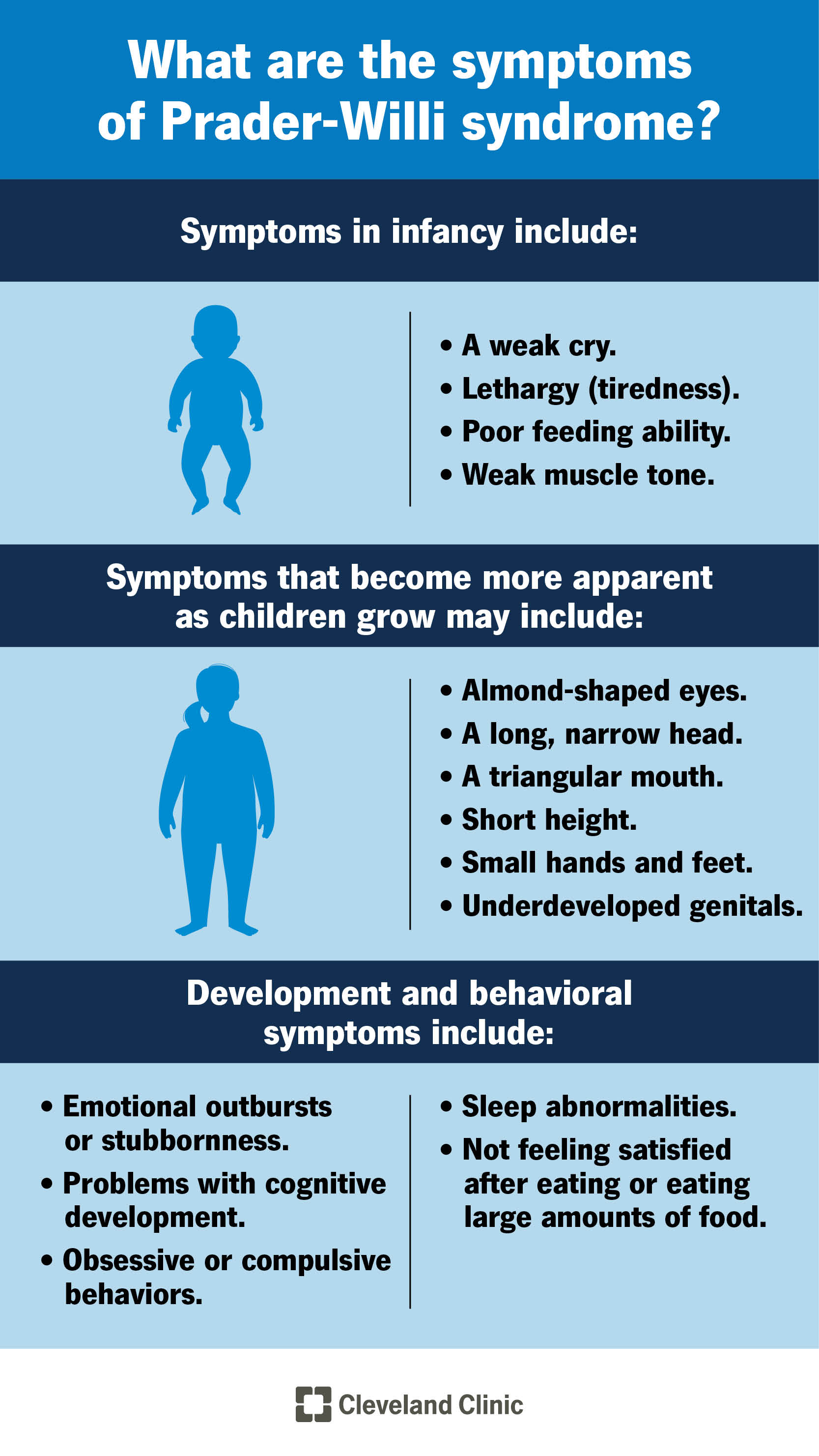
3. Clinical Features
PWS evolves in two major stages: infancy and childhood/adulthood.
Other common findings:
Behavioral and emotional issues: temper tantrums, stubbornness, obsessive–compulsive traits
Cognitive function: mild to moderate intellectual disability
Endocrine abnormalities: growth hormone deficiency, hypothyroidism, adrenal insufficiency
Sleep disturbances: sleep apnea, excessive daytime sleepiness
Skeletal: scoliosis, osteoporosis (due to low GH and sex hormones)
4. Diagnosis
Diagnostic approach includes:
Genetic testing: DNA methylation analysis confirms the loss of paternal gene expression at 15q11–q13 (gold standard).
Chromosomal microarray or FISH: to identify deletions or UPD.
Clinical suspicion: based on hypotonia, feeding issues, and later hyperphagia with obesity.
5. Management
There is no cure, but multidisciplinary management dramatically improves quality of life.
1. Endocrine and Hormonal Therapy
Growth hormone (GH) replacement: improves growth, muscle mass, metabolism, and body composition.
Sex hormone replacement: supports puberty and bone health.
Monitor thyroid and adrenal function regularly.
2. Nutritional and Weight Management
Controlled diet and calorie restriction from early childhood
Family supervision of food access due to compulsive eating behavior
Regular exercise programs
3. Behavioral and Developmental Support
Early physical, speech, and occupational therapy
Behavioral therapy to manage emotional outbursts and obsessive tendencies
Educational support for cognitive delay
4. Medical Monitoring
Screening for sleep apnea, scoliosis, diabetes, and obesity-related complications
6. Prognosis
Life expectancy is reduced mainly due to obesity-related complications (diabetes, heart disease, sleep apnea).
With early diagnosis, GH therapy, and lifestyle management, patients can live into adulthood with improved independence.
Ongoing family and social support are essential.
7. Summary Table: Prader–Willi Syndrome Overview
8. Conclusion
Prader–Willi syndrome is a prime example of how genetic imprinting errors can profoundly influence growth, metabolism, and behavior.
Early recognition—especially in infants with unexplained hypotonia and feeding issues—allows timely intervention.
With structured hormonal therapy, weight control, and behavioral support, individuals with PWS can achieve better health, independence, and quality of life.
Comment